MuTect2 Pitfalls¶
The standard parameters in MuTect2 (GATK v3.7) are very strict. For example, a somatic variant (no matter its frequency in the tumor sample) is discarded if the variant is seen in more than one read in the normal sample. This is particularly problematic for panel sequencing with read depths >1000X in targeted regions.
We propose solving this issue by raising the values of two MuTect2 parameters as indicated in the table below. By setting max_alt_alleles_in_normal_count to a very high number (there is no option to completely disable this filter), we never discard variants based on the absolute count of ALT alleles in the normal sample. Furthermore, we allow up to 10% of the normal reads to contain the ALT allele.
| Parameter | Default | Proposed |
|---|---|---|
| max_alt_alleles_in_normal_count | 1 | 10000000 |
| max_alt_allele_in_normal_fraction | 0.03 | 0.10 |
These more relaxed settings inevitable lead to an increased number of false positives. We filter those using custom filters (described below).
Motivation¶
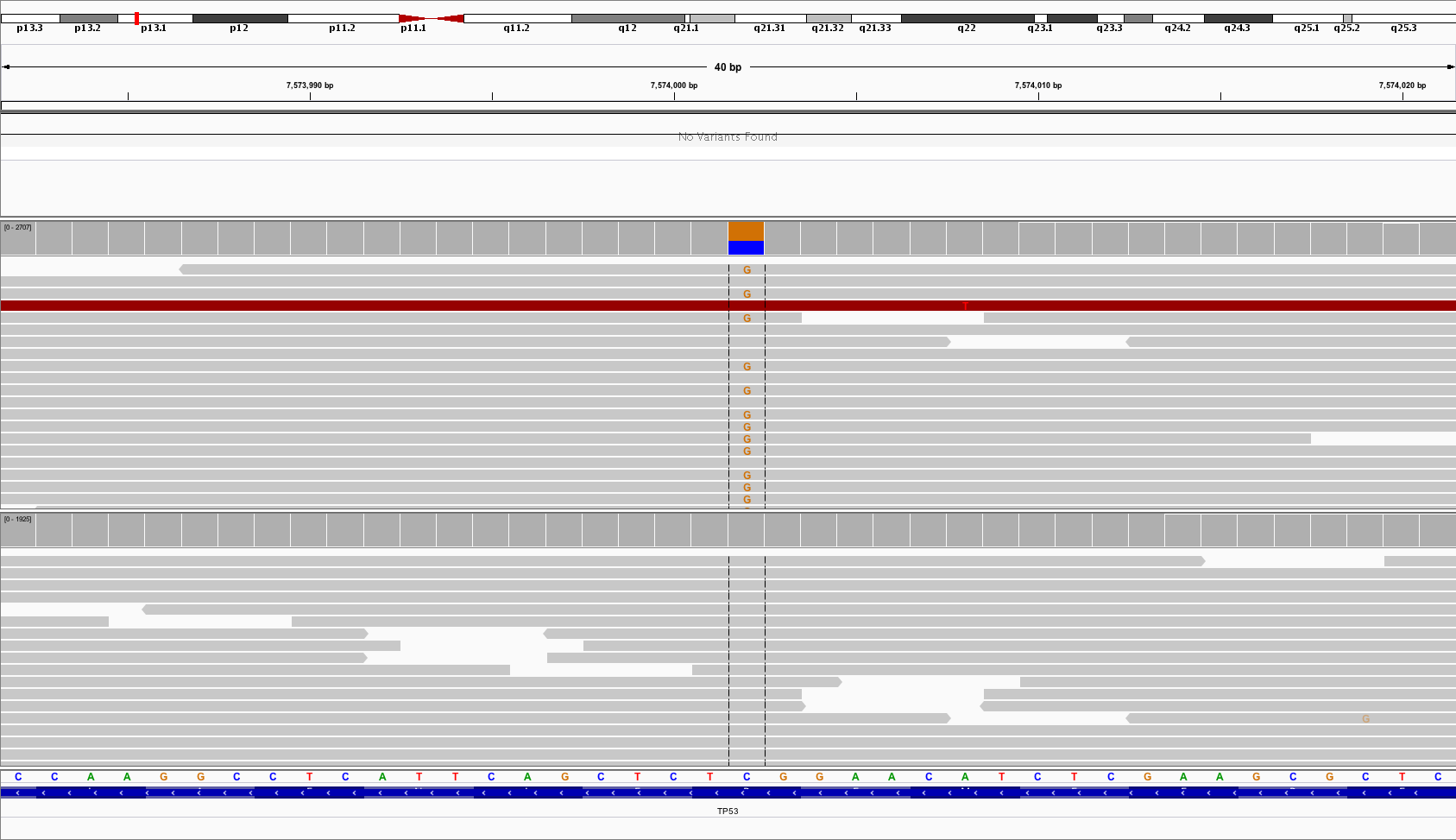
The IGV screenshot below shows an example of a variant in TP53 which is not called by MuTect2 with default parameters. The allele frequency in the tumor sample is 58% (1528/2634). However, the variant is filtered out by MuTect2, because it fails both filters above. The allele frequency in the normal sample is 1.1% (21/1920).
Downstream Filters¶
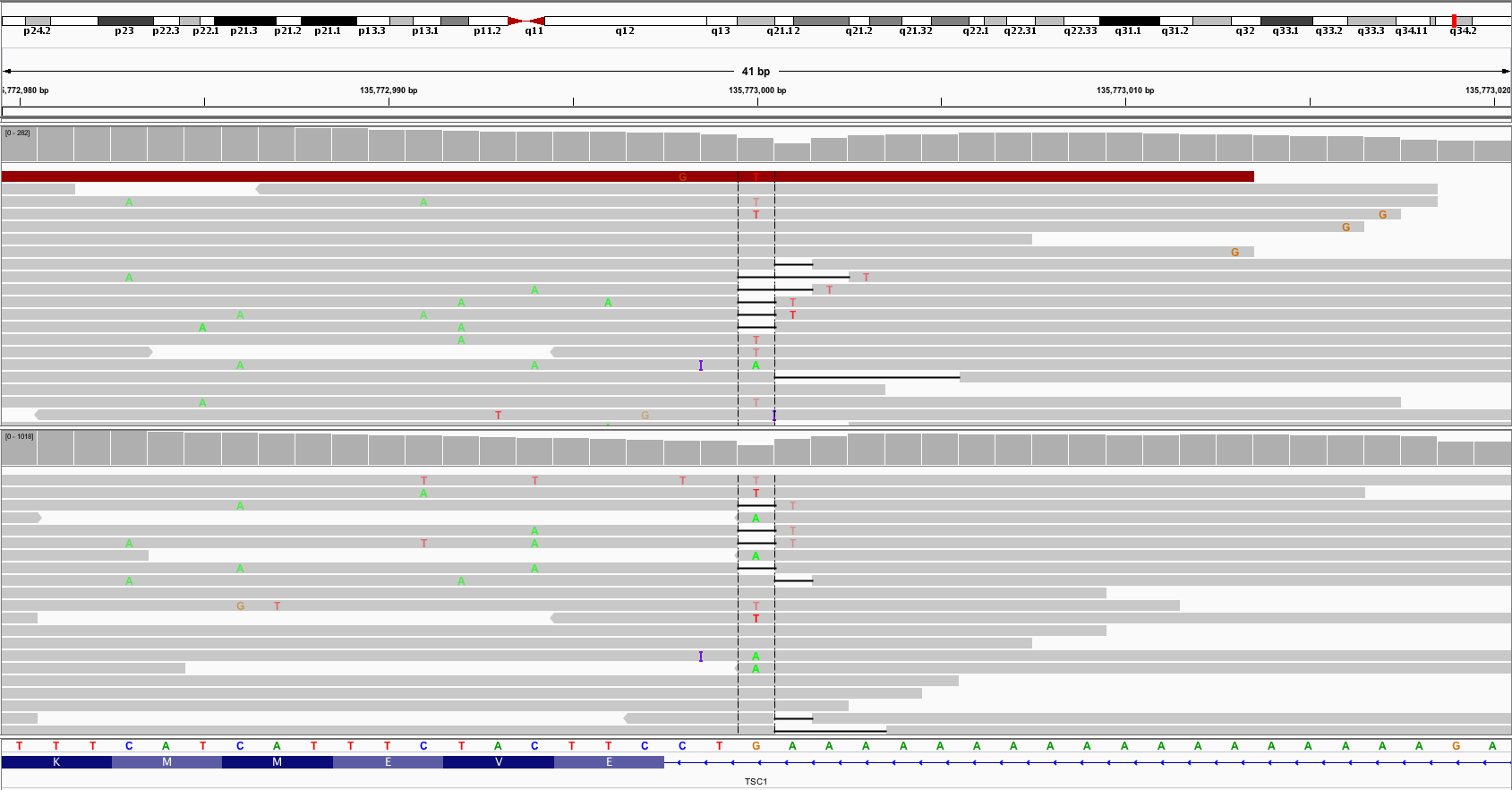
MuTect2 with standard parameters makes many false positive calls in noisy regions, and this only gets worse with the relaxed paramter settings. For example, the TSC1 variant in the screenshot below is not filtered by MuTect2, although it is clearly noise.
Another typical cause of false positives is similar allele frequencies in tumor and normal. MuTect2 will call variants in the tumor sample as somatic, as long as their frequencies in the normal sample are not above the 10% cut-off. In some cases, we therefore end up calling variants with higher allele frequencies in the normal than in the tumor as somatic.
To deal with the issue of false positives, we propose the following two metrics calculated from info in the MuTect2 VCF files:
We have found that filtering samples not meeting these three criteria:
effectively eliminates all false positives and never removes genuine variants.
The rationale behind these values is as follows: The QSS score is the average base quality of variant bases. If this is below 20, the region is very noisy, and the variant is not likely to be genuine. Furthermore, note that \(S_{AF} > 0.75\) is equivalent to \(AF_{tumor} > 4\cdot AF_{normal}\), which means that a variant with a “high” frequency in the normal sample is not called as somatic, unless its frequency in the tumor sample is “much” higher.
Adding Second Variant Caller¶
Despite the optimization of MuTect2 described above, important variants may still be missed. The screenshot below shows a clear hotspot variant (BRAF V600E) filtered out by MuTect2. This particular variant has been independently validated using a complimentary method.

In fact, MuTect2 not only filters out V600E but also a variant in the neighboring codon. The explanation for both variants being filtered is indicated as clustered_events and multi_event_alt_allele_in_normal in the VCF file.
Simply disabling these filters leads to an explosion in false positive calls. Instead, we have found that using an additional variant caller besides MuTect2 may rescue erroneously filtered variants. More precisely, we run VarScan2 and check if any high confidence variants were also called (but subsequently filtered) by MuTect2. If so, we keep these in a separate file for manual inspection. In the particular example above, both BRAF variants are rescued with no extra false positives added.